Input File Format¶
“Skeleton-RHF”¶
Here, we show an example of a “skeleton-RHF” input file used for the RHF energy calculation of one water molecule with the 6-31G(d) basis sets:
6-31G*
0 3
O 10.438 -22.339 1.220
H 11.395 -22.339 1.220
H 10.198 -21.412 1.220
The input file format is schematically written by
Name_of_Basis_Sets
Molecular_Charge Number_of_Atoms
Atomic_Name X Y Z
Atomic_Name X Y Z
Atomic_Name X Y Z
...
| Line | Column | Description | Acceptable Variables | Note |
|---|---|---|---|---|
| 1 | 1 | Name of Basis Sets | sto-3g, 6-31g, 6-31g*, 6-31g** | Both capital and lower-case letters are O.K. |
| 2 | 1 | Molecular charge | Integer values | Only closed-shell |
| 2 | 2 | Number of atoms of molecule | Integer values | |
| 4- | 1 | Atomic name | Up to Third-row Atoms (namely, H - Ar) | Both capital and lower-case letters are O.K. |
| 4- | 2-4 | Cartesian coordinates | Floating-point numbers | In Angstrom |
OpenFMO¶
Introduction¶
OpenFMO adopts the same input-file format as the FMO calculations implemented in GAMESS ab initio quantum chemistry package. Since some of the input groups used in GAMESS are directory used in the OpenFMO, the GAMESS documentations, Input Description and Further Information , are also useful for the users of OpenFMO. Table 3 lists the input groups used in OpenFMO. Both lower-case and capital letters can be used to write the input groups and their options in the input files for OpenFMO.
| name | function | options |
|---|---|---|
| $CONTRL | chemical control data | maxit, nprint, itol, icut |
| $BASIS | basis set | ngauss, gbasis, ndfunc, npfunc |
| $INTGRL | two-electron integrals | nintmx, nintic |
| $GDDI | MPI dynamic process management | ngroup, niogroup, nioprocs |
| $SCF | SCF control | diis, npunch, conv |
| $FMOPRP | FMO convergers | sccconv, maxscc |
| $FMOXYZ | atomic coordinates for FMO | Name, ZNUC, X, Y, Z |
| $FMO | Define FMO fragments | nfrag, icharg, indat |
| $FMOLMO | localized MO for FMO boundaries | Refer to “HMOs.txt” distributed by GAMESS |
| $FMOBND | FMO bond cleavage definition |
Simple Example (Glycylglycine)¶
Here, we show an example of an OpenFMO input file used for the FMO-RHF/STO-3G energy calculation of one glycylglycine. Figure 2 illustrates the structure and its fragmentation points:
- Fragment 1 includes N1, H2, H3, C4, H5, and H6 atoms
- Fragment 2 includes C7, O8, N9, H10, C11, H12, and H13 atoms.
- Fragment 3 includes C14, O15, O16, and H17 atoms.
- The alpha-carbon atoms (C4 and C11) are treated as bond-detached atoms (BDAs).
- Thus, the detached bonds are assigned to the atoms on the other side (C7 and C14), which are called bond-attached atoms (BAAs).
$FMOBND group in the input file tells OpenFMO program the way of the bond detachments. In the input file, the number of server groups (See Figure 1) is set to 1 through the niogroup option in $GDDI group. As mentioned in Command Line Options, the command-line argument -niogroup can also set up the number of server groups. Similarly, the command-line argument -nioprocs can also set up the size of each server groups, which is defined by the nioprocs option in $GDDI group.
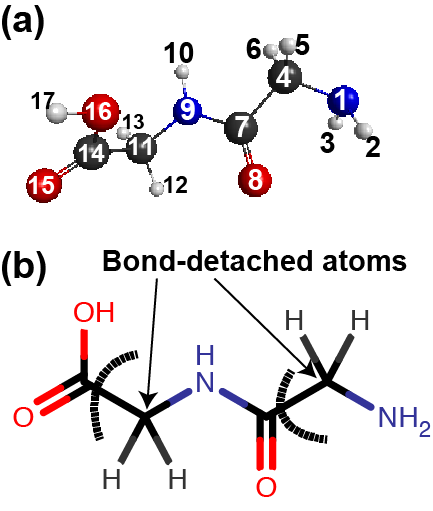
Figure 2. Glycylglycine: (a) Structure and Atom Numbering (b) Fragmentation
$gddi niogroup=1 nioprocs=1 $end
$fmo nfrag=3 ICHARG(1)= 0,0,0
INDAT(1)=0, 1, -6, 0, 7, -13, 0, 14, -17,0 $end
$basis gbasis=sto ngauss=3 $end
$fmoxyz
N 7.0 3.5584 0.0170 0.1638
H 1.0 3.6446 -0.8687 -0.3332
H 1.0 3.4912 -0.2124 1.1546
C 6.0 2.3540 0.7121 -0.2674
H 1.0 2.2350 1.6486 0.2858
H 1.0 2.4304 0.9444 -1.3339
C 6.0 1.1558 -0.1725 0.0097
O 8.0 1.1192 -0.9807 0.9350
N 7.0 0.1194 0.0665 -0.8809
H 1.0 0.2322 0.7805 -1.5946
C 6.0 -1.1505 -0.6217 -0.8231
H 1.0 -0.9953 -1.6290 -0.4254
H 1.0 -1.5383 -0.6729 -1.8442
C 6.0 -2.1620 0.0850 0.0422
O 8.0 -3.2962 -0.3376 0.2304
O 8.0 -1.6980 1.2320 0.5903
H 1.0 -2.3743 1.6726 1.1478
$end
$FMOLMO
STO-3G 5 5
1 0 -0.117784 0.542251 0.000000 0.000000 0.850774
0 1 -0.117787 0.542269 0.802107 0.000000 -0.283586
0 1 -0.117787 0.542269 -0.401054 -0.694646 -0.283586
0 1 -0.117787 0.542269 -0.401054 0.694646 -0.283586
0 1 1.003621 -0.015003 0.000000 0.000000 0.000000
$end
$FMOBND
-4 7 STO-3G
-11 14 STO-3G
$end
$CONTRL Group¶
| Option | Acceptable Variables | Explanation | Note |
|---|---|---|---|
| maxit = | positive integer | Maximum number of SCF iteration cycles | default: maxit = 30 |
| nprint = | integer value | Print control flag | default: nprint = 0 |
| itol = | positive integer | primitive cutoff factor 10**(-n) | default: itol = 20 |
| icut = | positive integer | 2e-integral cutoff factor 10**(-n) | default: icut = 12 |
$BASIS Group¶
OpenFMO can treat minimum and double-zeta Gaussian basis functions of up to third-row atoms, including STO-3G, 6-31G, 6-31G(d) and 6-31G(d,p).
| Option | Acceptable Variables | Explanation | Note |
|---|---|---|---|
| ngauss = | 3 (for STO-3G) or 6 (for 6-31G) | the number of Gaussians | default: ngauss = 3 |
| gbasis = | sto (STO-3G) or n31 (for 6-31G) | basis function | default: gbasis=sto |
| ndfunc = | 0 (for STO-3G and 6-31G) or 1 ( for 6-31G* and 6-31G**) | number of heavy atom polarization functions to be used | default: ndfunc = 0 |
| npfunc = | 0 (for STO-3G, 6-31G, and 6-31G*) or 1 (for 6-31G**) | number of light atom, p type polarization | default: npfunc = 1 |
$INTGRL Group¶
| Option | Acceptable Variables | Explanation | Note |
|---|---|---|---|
| nintmx = | positive integer | Maximum no. of integrals in a record block | default: nintmx = 15000 |
| nintic = | 0 or positive integer | buffer size / processes in MB | default: nintic = 512; See the command-line argument -B in Command Line Options. |
$GDDI Group¶
$GDDI group tells the OpenFMO program how to manage the MPI dynamic processes in FMO calculations. However, as described in Command Line Options, users had better use the command-line options, -ng, -niogroup, and -nioprocs rather than $GDDI group in the input file in order to manage the MPI dynamic processes.
| Option | Acceptable Variables | Explanation | Note |
|---|---|---|---|
| ngroup | positive integer | Number of worker groups | default: ngroup = 1; OpenFMO prefers this option to the -ng command-line argument |
| niogroup | positive integer | Number of server groups | default: niogroup=1; OpenFMO prefers this option to the -niogroup command-line argument |
| nioprocs | positive integer | Size of each server group | default: nioprocs=1; OpenFMO prefers this option to the -nioprocs command-line argument |
$SCF Group¶
| Option | Acceptable Variables | Explanation | Note |
|---|---|---|---|
| diis = | false or true | Pulay’s DIIS interpolation | default: diis=true |
| npunch = | 0, 1, or 2 | option for output | |
| conv = | positive integer | SCF density convergence criteria 10**(-n) | default: conv=7 |
$FMOPRP Group¶
| Option | Acceptable Variables | Explanation | Note |
|---|---|---|---|
| sccconv = | positive integer | monomer SCF energy convergence criterion; 10**(-n) | default: sccconf = 7 |
| maxscc = | positive integer | the maximum number of monomer SCF iterations | default: maxscc=30 |
$FMOXYZ Group¶
$FMOXYZ group defines the molecular data as follows:
$fmoxyz
Name ZNUC X Y Z
Name ZNUC X Y Z
Name ZNUC X Y Z
...
$end
- NAME = atomic name
- ZNUNC = nuclear charge
- X, Y, Z = Cartesian coordinates in Angstrom
$FMO Group¶
| Option | Acceptable Variables | Explanation | Note |
|---|---|---|---|
| nfrag = | positive integer | number of distinct fragments present | default: nfrag = 1 |
| icharg(1) = | an array of positive integers | an array of charges on the fragments | default: all 0 charges |
| indat(1) = | Described below | an array assigning atoms to fragments |
The indat option in $FMO group tells OpenFMO program an array assigning atoms to fragments. We would like to cite its explanation from the GAMESS documentation, Input Description :
INDAT(i)=m assigns atom i is to fragment m.INDAT(i) must be given for each atom.An element is one of the following:I or I -Jwhere I means atom I, and a pair I, -J meansthe range of atoms I-J. There must be no spaceafter the “-“!Example:indat(1)=1,1,1,2,2,1 is equivalent toindat(1)=0, 1,-3,6,0, 4,5,0Both assign atoms 1,2,3 and 6 to fragment 1,and 4,5 to fragment 2.
$FMOBND Group¶
The atom indices involved in the bond detachment are given, in pairs for each bond. Bonds are always detached between fragments.
$fmobnd
-I1 J1 NAM1
-I2 J2 NAM2
-I3 J3 NAM3
...
$end
I and J are positive integers giving absolute atom indices. NAMs are hybrid orbital set names, defined in $FMOLMO group. Each line is allowed to have different set of NAMs, which can happen if different type of bonds are detached.
Note that OpenFMO code definitely reads $FMOBND group. If your FMO calculation does NOT need any bond detachement, its input file has to include a blank line sandwiched between $fmobnd and $end keywords (see subsection TCNE-(Benzene)8-TCNE in Samples.):
$fmobnd
$end
$FMOLMO or $FMOHYB Group¶
Hybrid orbitals are used to describe bond detachment when dividing a molecule into fragments. These are the familiar sp3 orbitals for Carbon atom, plus the 1s core orbital.
OpenFMO can treat STO-3G, 6-31G, 6-31G(d), and 6-31G(d,p) basis sets. Therefore, you can extract the orbitals (NAM=STO-3G, 6-31G, 6-31G*, or 6-31G**) to be put into $FMOLMO group taken from “HMOs.txt” distributed by GAMESS.
Note that OpenFMO code definitely reads $FMOLMO or $FMOHYB group. Even if your FMO calculation does NOT need any bond detachement, you have to write some hybrid molecular orbitals (HMOs) taken from “HMOs.txt” for $FMOLMO/$FMOHYB group in its input file. However, the HMOs are anything O.K. and the OpenFMO code does not use them in the FMO calculation. See subsection TCNE-(Benzene)8-TCNE in Samples.