Samples¶
Introduction¶
In this section we show the results of several test calculations obtained from the “skeleton-RHF” and OpenFMO codes. To this end, we used the ACC nodes of System B of Institute for Solid State Physics (ISSP) supercomputer at the University of Tokyo; the configuration of ACC node is as follows:
- CPU: 2 Intel Xeon E5-2680 v3 2.5GHz (12core * 2)
- GPU: 2 nVIDIA Tesla K40 ( 2880 CUDA core * 2)
- MEM: DDR4-2133 128GB
- FDR InfiniBand
The compilers and libraries used are as follows:
- icc version 16.0.4 (gcc version 4.8.5 compatibility)
- mpiicc for the Intel(R) MPI Library 5.1.3 for Linux*
- Cuda compilation tools, release 7.0, V7.0.27
Glycylglycine¶
We again treat the FMO-RHF/STO-3G
calculation of one glycylglycine
illustrated in Figure 2,
whose input file is explained
in Simple Example (Glycylglycine).
You can download the input (digly-ofmo-rhf.inp),
output (digly-ofmo-rhf.out), and
PBS job (job.sh) files used.
For the calculation,
we used one ACC node
with 1 data server of 1 rank and 2 worker groups of 1 rank.
We also performed the RHF/STO-3G calculation
with “skeleton-RHF” code on the same glycyglycine.
You can download the input (digly-rhf.inp),
output (digly-rhf.inp), and
PBS job (job2.sh) files used.
For the calculation,
we performed 24 threads execution of “skeleton-RHF” code
with one GPU unit.
We can see the FMO-RHF/STO-3G calculation reproduces the energy of the molecule obtained from the RHF/STO-3G calculation, as listed in Table 11
| RHF | FMO |
|---|---|
| -483.23779373 | -483.23772776 |
ala10 in alpha-helix conformation¶
We demonstrate the FMO-RHF/6-31G(d)
calculation of the alanine polypeptide
in an ideal alpha-helix conformation (called ala10 hereafter)
plotted in Figure 3.
The alpha-carbon atoms,
as marked by the arrows in Figure 3 (b),
are treated as bond-detached atoms (BDAs).
The coordinate is taken from the previous study [NA11].
The input (ala10-ofmo-rhf.inp),
output (ala10-ofmo-rhf.out), and
PBS job (job3.sh) files used can be downloaded.
For the calculation,
we used one ACC node
with 1 data server of 1 rank and 2 worker groups of 1 rank.
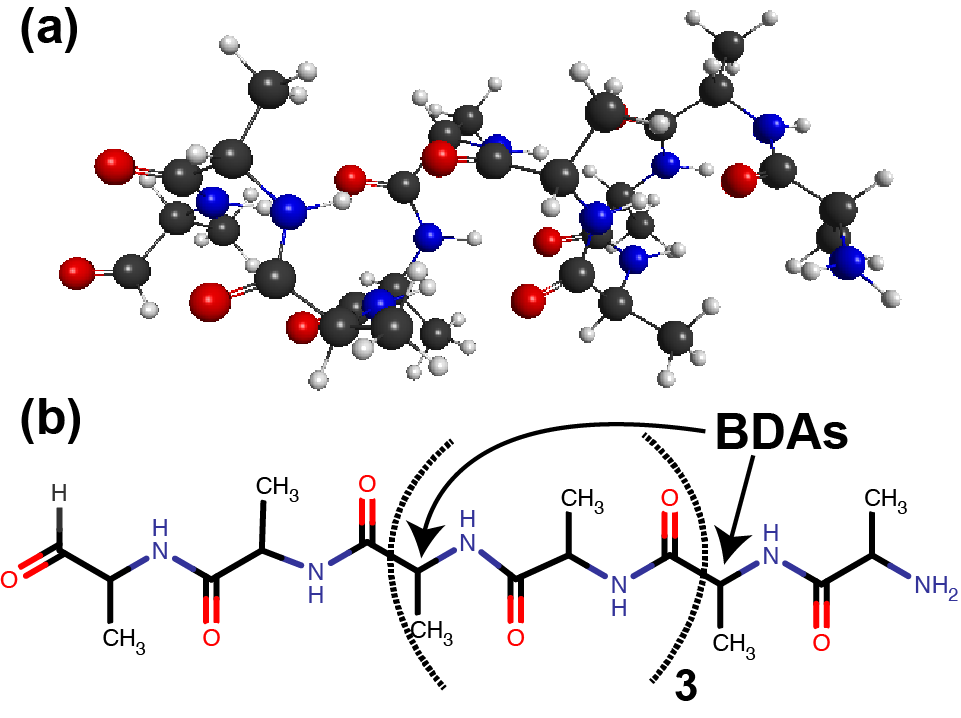
Figure 3. ala10: (a) Structure (b) Fragmentation
We also performed the RHF/6-31G(d) calculation
with “skeleton-RHF” code on the same ala10.
You can download the input (ala10-rhf.inp),
output (ala10-rhf.out), and
PBS job (job4.sh) files used.
For the calculation,
we performed 24 threads execution of “skeleton-RHF” code
with one GPU unit.
Table 12 compares
the resultant FMO-RHF/6-31G(d) energy with
the RHF/6-31G(d) one.
Table 12 also lists
the RHF/6-31G(d) and FMO-RHF/6-31G(d) energies of ala10
calculated with GAMESS;
you can download the input files (
Ala10AlphaRhfN31d.inp
and
Ala10AlphaFmoRhfN31d.inp)
used for GAMESS.
| OpenFMO | GAMESS | ||
|---|---|---|---|
| RHF | FMO | RHF | FMO |
| -2459.51658600 | -2459.51865445 | -2459.51658597 | -2459.51863956 |
TCNE-(Benzene)8-TCNE¶
We, here, demonstrate a FMO-RHF/6-31G(d,p) calculation
of the model system TCNE-(Benzene)8-TCNE,
where eight perfectly eclipsed-stack benzene molecules
are sandwiched by tetracyanoethylene (TCNE),
as plotted in Figure 4.
You can download the input
(TcneBenzen8Tcne-ofmo-rhf.inp),
output
(TcneBenzen8Tcne-ofmo-rhf.out), and
PBS job (job5.sh) files.
The coordinate is taken from the previous study [NA11].
The FMO calculation treats
each isolated molecule as a fragment,
thereby involving no bond detachement.
Note that the input file for OpenFMO has to define
$FMOBND and $FMOLMO/$FMOHYB groups if its FMO calculation involves no bond detachment ,
as TcneBenzen8Tcne-ofmo-rhf.inp;
in that case, the OpenFMO code does NOT use the hybrid molecular orbitals
defined by $FMOLMO/$FMOHYB group in the calculation and read
the blank line defined by $FMOBND group.
See $FMOLMO or $FMOHYB Group and $FMOBND Group.
For the calculation,
we used 8 ACC nodes
with 1 data server of 1 rank and 6 worker groups of 1 rank.
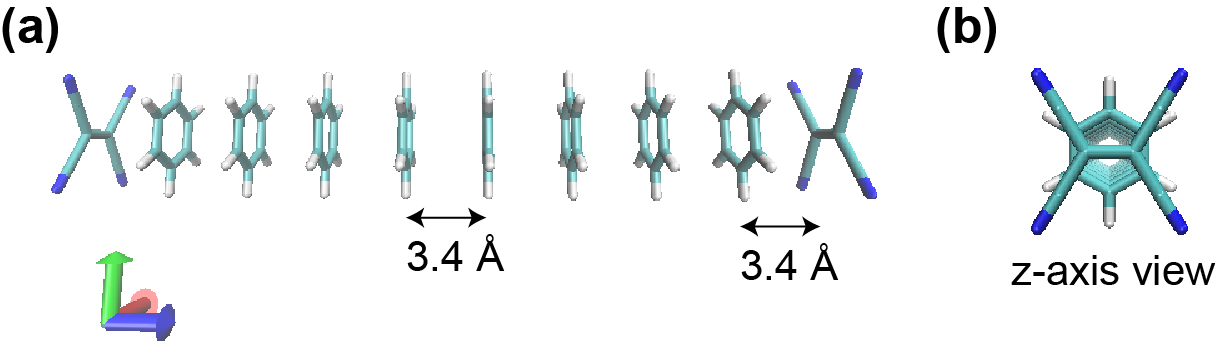
Figure 4. Structure of TCNE-(Benzene)8-TCNE possessing D2h molecular symmetry
In addition,
You can download the input (TcneBenzen8Tcne-rhf.inp),
output (TcneBenzen8Tcne-rhf.out), and
PBS job (job6.sh) files
used for the RHF/6-31G(d,p) calculation
with “skeleton-RHF” code.
Table 13 compares
the resultant FMO-RHF/6-31G(d,p) energy with
the RHF/6-31G(d,p) one.
| RHF | FMO |
|---|---|
| -2735.45391614 | -2735.45313547 |
DNA¶
This subsection shows an example of the FMO-RHF/6-31G(d) calculation
of dephosphorilated GTTTG B-DNA oligomer (dGTTTG) neutralized by
8 Na+ ions,
as illustrated in Figure 5 (a).
You can download the input
(dna-ofmo-rhf.inp),
output
(dna-ofmo-rhf.out), and
PBS job (job7.sh) files.
We constructed the structure of dGTTTG
by using the NBA program
from AmberTools
package in line with the previous study [RP14].
Figure 5 (b) shows how to place Na+ ion,
which follows the previous study [FWK+14].
Figure 5 (c) shows that the FMO calculation
treats the cabon atoms at the 5’ and
4’ positions as BDA (bond-detached atom) and BAA (bond-attached atom),
respectively.
For the calculation,
we used 8 ACC nodes
with 1 data server of 1 rank and 6 worker groups of 1 rank.
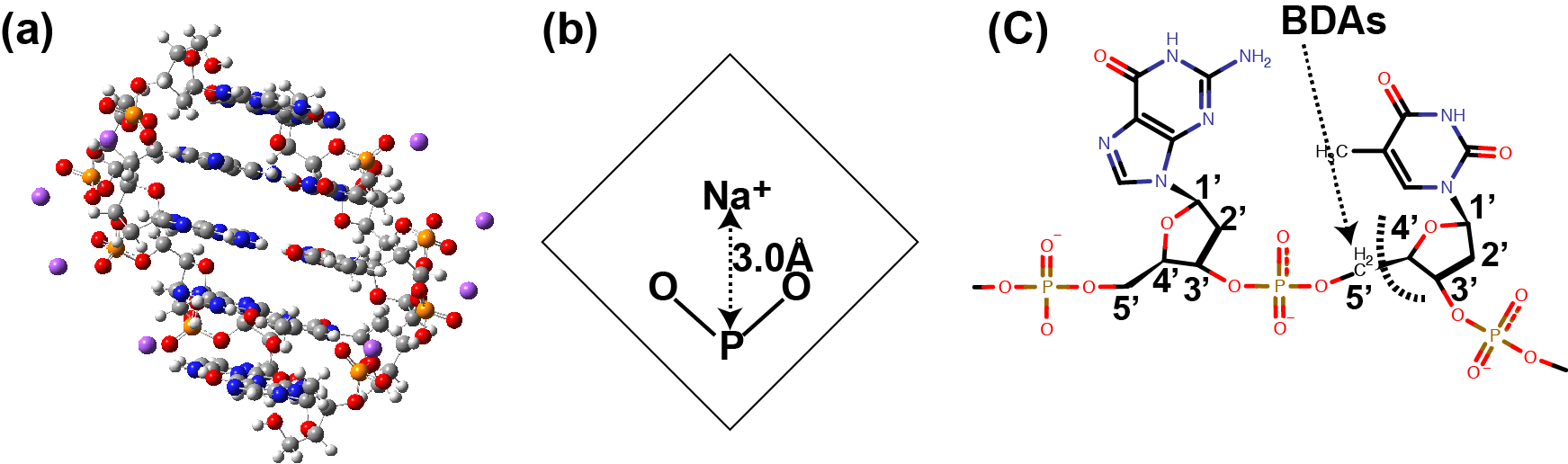
Figure 5. dGTTTG with 8 Na+ ions: (a) Structure, (b) Placement of Na+ ion, and (c) Fragmentation
For comparison, we performed the same calculation using OpenFMO
turning off its GPU acceleration;
you can see the detail of the results in the output
file dna-ofmo-rhf-offgpu.out.
Table 14 lists the total SCF energy and
computational time obtained using OpenFMO with/without GPU units.
Note that the computational times taken from the output files,
which are printed as “total etime = …”, are crude,
but are probably useful information in this test calculation.
We can confirm that the usage of GPU units does NOT affect the resultant SCF energy.
The GPU-accelerated OpenFMO halves the computational time
calculated with the OpenFMO
even though the system size is modest and
the computational settings, such as the types of the two-electron integral,
are not optimized.
| SCF Energy | Computational Time | |
|---|---|---|
| CPU + GPU | -14004.75470797 | 1049.715826 |
| Only CPU | -14004.75470797 | 2154.053616 |
| [FWK+14] | Kaori Fukuzawa, Chiduru Watanabe, Ikuo Kurisaki, Naoki Taguchi, Yuji Mochizuki, Tatsuya Nakano, Shigenori Tanaka, and Yuto Komeiji. Accuracy of the fragment molecular orbital (fmo) calculations for dna: total energy, molecular orbital, and inter-fragment interaction energy. Comput. Theor. Chem., 1034:7 – 16, 2014. URL: http://www.sciencedirect.com/science/article/pii/S2210271X14000577, doi:https://doi.org/10.1016/j.comptc.2014.02.002. |
| [NA11] | (1, 2) Hirotaka Nishioka and Koji Ando. Electronic coupling calculation and pathway analysis of electron transfer reaction using ab initio fragment-based method. i. fmo–lcmo approach. J. Chem. Phys., 134(20):204109, 2011. URL: http://dx.doi.org/10.1063/1.3594100, arXiv:http://dx.doi.org/10.1063/1.3594100, doi:10.1063/1.3594100. |
| [RP14] | Pablo Ramos and Michele Pavanello. Quantifying environmental effects on the decay of hole transfer couplings in biosystems. J. Chem. Theory Comput., 10(6):2546–2556, 2014. URL: https://doi.org/10.1021/ct400921r, arXiv:https://doi.org/10.1021/ct400921r, doi:10.1021/ct400921r. |